Workflow
One approach for identifying splicing variants is to use both genome and transcriptome data and perform some statistical associations study between the genomic variants and transcriptome changes. There are many studies in this regard (Jung et al., Nature Genetics, 2015; Shiraishi et al., Genome Research, 2018; PCAWG Transcriptome Core Group et al., Nature, 2020). However, we need both genome and transcriptome sequence data for using this approach, and such cases are not so common other than very organized projects like TCGA. On the other hand, in Sequence Read Archive, there are hundreds of thousands of publicly available transcriptome sequence data, and several studies make the most of this massive resource, such as recount3. So, the question is can we identify splicing variants just using transcriptome sequence data?
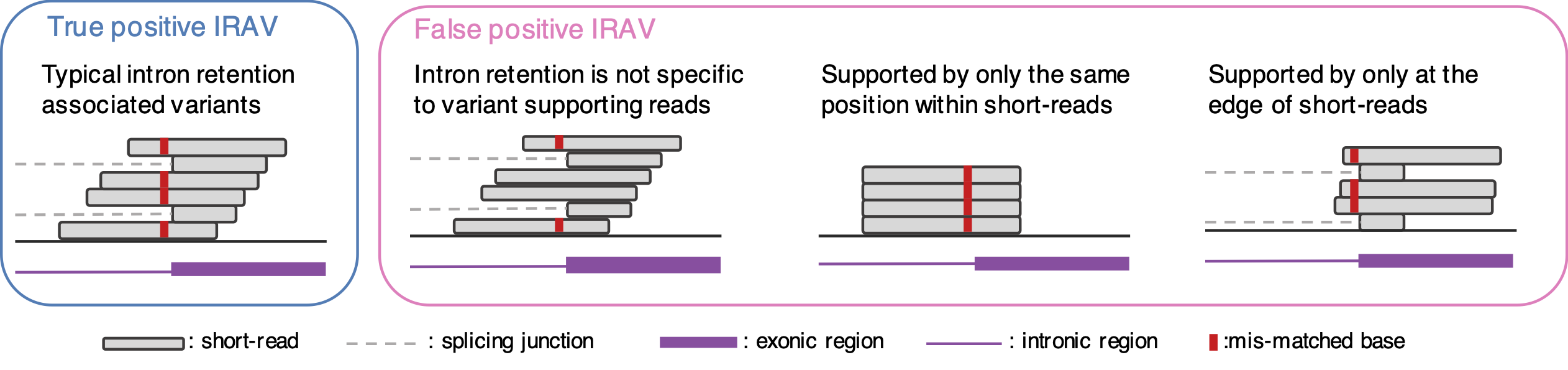
In fact, there is a particular type of splicing variant which could be detected just using transcriptome sequencing data. That is genomic variants causing intron retention (intron retention associated variants, IRAVs). As Figure 1 shows, when a variant causes intron retention, that variant will remain in the retained reads. So it seems that using this phenomenon, we may be able to identify intron retention associated variants only using transcriptome sequencing data.
Here is the idea of our proposed method to identify IRAVs. We start with aligned BAM files. First, we list up the position where mismatch bases are accumulated (we confine three exonic bases and six intronic bases for donor sites, and one exonic base and six intronic bases for acceptor sites). Then, we check whether most of the intron retention reads contain mismatch bases or not (since if the intron retention is not specific to variant supporting reads, then this variant may not be associated with intron retention). Moreover, we removed other types of false positives (Figure 2). If all the conditions are OK, we call the variant as IRAVs. Please note that we removed the common variants (AF >= 0.01 by gnomAD), because these are not usually related to disease, while these tend to be the source of false positives.

Pathogenecity
To identify IRAVs related to disease, we focused on the positional relationship between pathogenic variants registered in ClinVar.
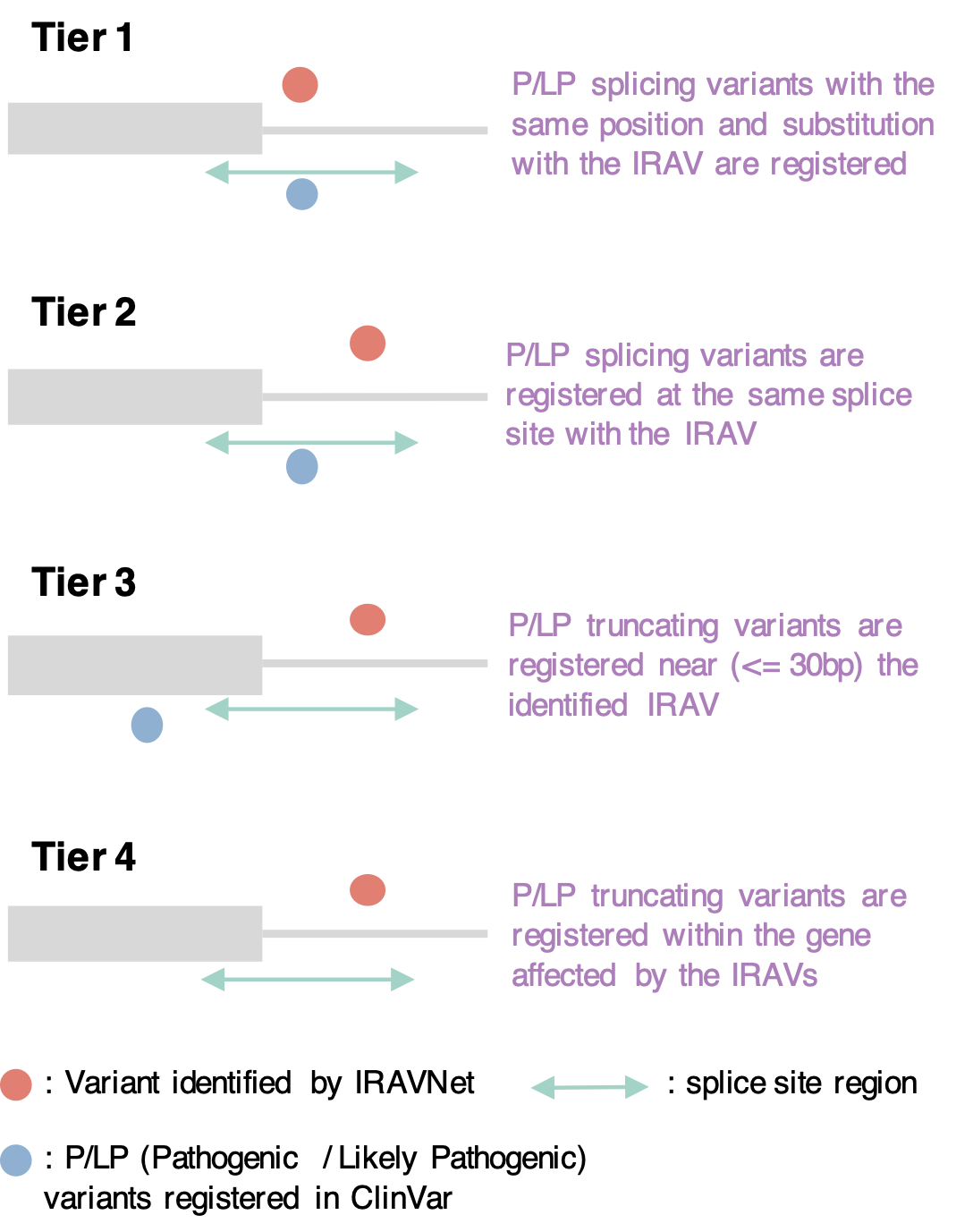
We extracted pathogenic IRAVs using five-tier model.
First, for an IRAV, if a pathogenic variant with the same position and substitution is registered in ClinVar, this IRAV is defined as Tier1 pathogenic IRAV.
Next, if pathogenic variants sharing the same splice site with the IRAV are registered, this IRAV is classified into Tier2.
When a pathogenic variant is observed near the IRAV (within 30 bp at the transcript level), this IRAV is deemed Tier3.
Finally, when there is a pathogenic truncating variant within the gene affected by the IRAV, then this IRAV is set to Tier4.
All the other IRAVs are classified as Tier5.
Main Contributer
Ai Okada
Yuichi Shiraishi
Citation
Please site the following paper when you use the resource in this site.
Shiraishi et al., Systematic identification of intron retention associated variants from massive publicly available transcriptome sequencing data, Nat Commun., 2022 Sep 29;13(1):5357
.
Acknowledgment
This work is supported by Grant-in-Aid for Scientific Research (KAKENHI 18H03327, 21H03549) and Grand-in-Aid from the Japan Agency for Medical Research and Development (Platform Program for Promotion of Genome Medicine: 20km0405207h9905, Program for an Integrated Database of Clinical and Genomic Information: 20kk0205014h0005, Practical Research Project for Rare / Intractable Diseases: 20ek0109485h0001).
Notes
Although we performed screening of IRAVs on TCGA transcriptome data as well as SRA, we omitted those from TCGA. The current algorithm cannot discern germline and somatic variants, and we suspect there might be some possibility that it may lead to the identification of an individual.
Database version and date
ClinVar: 2023-02-26
Cancer Gene Census: 2023-03-05
ACMG SF Gene: v3.0
ClinGen Dosage Sensitivity: 05 Mar, 2023